Genomik
Genomics
Genomik adalah bidang biologi interdisipliner yang berfokus pada struktur, fungsi, evolusi, pemetaan, dan pengeditan genom. Genom adalah rangkaian lengkap DNA organisme, termasuk semua gennya. Berbeda dengan genetika, yang mengacu pada studi tentang gen individu dan peran mereka dalam pewarisan, genomik bertujuan untuk karakterisasi kolektif dan kuantifikasi semua gen organisme, keterkaitannya dan pengaruhnya terhadap organisme. Gen dapat mengarahkan produksi protein dengan bantuan enzim dan molekul kurir. Pada gilirannya, protein membentuk struktur tubuh seperti organ dan jaringan serta mengontrol reaksi kimia dan membawa sinyal antar sel. Genomik juga melibatkan sekuensing dan analisis genom melalui penggunaan sekuensing DNA dan bioinformatika tinggi untuk mengumpulkan dan menganalisis fungsi dan struktur seluruh genom. Kemajuan dalam genomik telah memicu revolusi dalam penelitian berbasis penemuan dan biologi sistem untuk memfasilitasi pemahaman tentang sistem biologis yang paling kompleks sekalipun seperti otak.
Bidang ini juga mencakup studi tentang fenomena intragenomik (dalam genom) seperti epistasis (efek satu gen pada gen lain), pleiotropi (satu gen mempengaruhi lebih dari satu sifat), heterosis (kekuatan hibrid), dan interaksi lain antara lokus dan alel dalam genom.
Daftar Isi
Sejarah
Etimologi
Dari bahasa Yunani ΓΕΝ gen , "gene" (gamma, epsilon, nu, epsilon) yang berarti "menjadi, ciptaan, kreasi, kelahiran ", dan varian berikutnya: silsilah, genesis, genetika, genik, genomere, genotipe, genus dll. Sedangkan kata genome (dari bahasa Jerman Genom , dikaitkan dengan Hans Winkler) digunakan dalam bahasa Inggris sejak tahun 1926, istilah genomik diciptakan oleh Tom Roderick, seorang ahli genetika di Laboratorium Jackson (Bar Harbor, Maine), sambil minum bir pada pertemuan yang diadakan saya n Maryland tentang pemetaan genom manusia pada tahun 1986.
Upaya pengurutan awal
Mengikuti konfirmasi Rosalind Franklin tentang struktur heliks DNA, publikasi James D. Watson dan Francis Crick tentang Struktur DNA pada tahun 1953 dan publikasi Fred Sanger tentang urutan asam amino insulin pada tahun 1955, urutan asam nukleat menjadi target utama ahli biologi molekuler awal. Pada tahun 1964, Robert W. Holley dan rekannya menerbitkan urutan asam nukleat pertama yang pernah ditentukan, urutan ribonukleotida dari RNA transfer alanin. Memperluas pekerjaan ini, Marshall Nirenberg dan Philip Leder mengungkapkan sifat triplet kode genetik dan mampu menentukan urutan 54 dari 64 kodon dalam percobaan mereka. Pada tahun 1972, Walter Fiers dan timnya di Laboratory of Molecular Biology of the University of Ghent (Ghent, Belgia) adalah orang pertama yang menentukan urutan gen: gen untuk mantel protein Bacteriophage MS2. Kelompok Fiers memperluas kerja protein mantel MS2 mereka, menentukan urutan nukleotida lengkap dari bakteriofag MS2-RNA (yang genomnya hanya mengkode empat gen dalam 3569 pasangan basa) dan virus Simian 40 masing-masing pada tahun 1976 dan 1978.
Teknologi pengurutan DNA dikembangkan
Selain pekerjaan penting tentang urutan asam amino insulin, Frederick Sanger dan rekan-rekannya memainkan peran kunci dalam pengembangan teknik pengurutan DNA yang memungkinkan pembentukan rangkaian proyek sekuensing genom. Pada tahun 1975, dia dan Alan Coulson menerbitkan prosedur pengurutan menggunakan DNA polimerase dengan nukleotida berlabel radiol yang dia sebut sebagai teknik Plus dan Minus . Ini melibatkan dua metode terkait erat yang menghasilkan oligonukleotida pendek dengan ujung 3 'yang ditentukan. Ini dapat difraksinasi dengan elektroforesis pada gel poliakrilamida (disebut elektroforesis gel poliakrilamida) dan divisualisasikan menggunakan autoradiografi. Prosedur ini dapat mengurutkan hingga 80 nukleotida sekaligus dan merupakan peningkatan besar, tetapi masih sangat melelahkan. Namun demikian, pada tahun 1977 kelompoknya mampu mengurutkan sebagian besar dari 5.386 nukleotida dari bakteriofag beruntai tunggal φX174, melengkapi genom berbasis DNA yang diurutkan penuh pertama. Penyempurnaan metode Plus dan Minus menghasilkan penghentian rantai, atau metode Sanger (lihat di bawah), yang menjadi dasar dari teknik sekuensing DNA, pemetaan genom, penyimpanan data, dan analisis bioinformatis paling banyak digunakan dalam penelitian seperempat abad berikutnya. Pada tahun yang sama Walter Gilbert dan Allan Maxam dari Universitas Harvard secara independen mengembangkan metode Maxam-Gilbert (juga dikenal sebagai metode kimia ) pengurutan DNA, yang melibatkan pembelahan preferensial DNA pada basis yang diketahui, lebih sedikit metode yang efisien. Untuk karya terobosan mereka dalam pengurutan asam nukleat, Gilbert dan Sanger berbagi setengah Hadiah Nobel tahun 1980 dalam bidang kimia dengan Paul Berg (DNA rekombinan).
Genom lengkap
Kemunculan teknologi ini menghasilkan intensifikasi yang cepat dalam cakupan dan kecepatan penyelesaian proyek pengurutan genom. Urutan genom lengkap pertama dari organel eukariotik, mitokondria manusia (16.568 bp, sekitar 16.6 kb), dilaporkan pada tahun 1981, dan genom kloroplas pertama menyusul pada tahun 1986. Pada tahun 1992, kromosom eukariotik pertama, kromosom III ragi bir Saccharomyces cerevisiae (315 kb) diurutkan. Organisme hidup bebas pertama yang diurutkan adalah Haemophilus influenzae (1,8 Mb) pada tahun 1995. Tahun berikutnya, konsorsium peneliti dari laboratorium di seluruh Amerika Utara, Eropa, dan Jepang mengumumkan penyelesaian urutan genom lengkap pertama eukariota, S. cerevisiae (12.1 Mb), dan sejak itu genom terus diurutkan dengan kecepatan yang tumbuh secara eksponensial. Per Oktober 2011, urutan lengkap tersedia untuk: 2.719 virus, 1.115 archaea dan bakteri, dan 36 eukariota, di mana sekitar setengahnya adalah jamur.
Sebagian besar mikroorganisme yang genomnya telah diurutkan secara lengkap bermasalah patogen, seperti Haemophilus influenzae , yang mengakibatkan bias yang nyata dalam distribusi filogenetiknya dibandingkan dengan luasnya keragaman mikroba. Dari spesies berurutan lainnya, sebagian besar dipilih karena mereka adalah organisme model yang dipelajari dengan baik atau dijanjikan akan menjadi model yang baik. Ragi ( Saccharomyces cerevisiae ) telah lama menjadi organisme model penting untuk sel eukariotik, sedangkan lalat buah Drosophila melanogaster telah menjadi alat yang sangat penting (terutama pada tahap awal pra-molekuler genetika). Cacing Caenorhabditis elegans adalah model sederhana yang sering digunakan untuk organisme multisel. Ikan zebra Brachydanio rerio digunakan untuk banyak studi perkembangan pada tingkat molekuler, dan tumbuhan Arabidopsis thaliana adalah organisme model untuk tumbuhan berbunga. Ikan buntal Jepang ( Takifugu rubripes ) dan ikan buntal hijau tutul ( Tetraodon nigroviridis ) menarik karena genomnya yang kecil dan padat, yang mengandung sangat sedikit DNA tanpa kode dibandingkan dengan kebanyakan spesies . Anjing mamalia ( Canis familiaris ), tikus coklat ( Rattus norvegicus ), tikus ( Mus musculus ), dan simpanse ( Pan troglodytes ) adalah hewan teladan yang penting dalam penelitian medis.
Draf kasar dari genom manusia diselesaikan oleh Proyek Genom Manusia pada awal tahun 2001, yang menimbulkan banyak keriuhan. Proyek ini, selesai pada tahun 2003, mengurutkan seluruh genom untuk satu orang tertentu, dan pada tahun 2007 urutan ini dinyatakan "selesai" (kurang dari satu kesalahan dalam 20.000 basis dan semua kromosom tersusun). Bertahun-tahun setelah itu, genom dari banyak individu lain telah diurutkan, sebagian di bawah naungan Proyek 1000 Genom, yang mengumumkan pengurutan 1.092 genom pada Oktober 2012. Penyelesaian proyek ini dimungkinkan oleh perkembangan yang lebih dramatis. teknologi pengurutan yang efisien dan membutuhkan komitmen sumber daya bioinformatika yang signifikan dari kolaborasi internasional yang besar. Analisis berkelanjutan atas data genom manusia memiliki dampak politik dan sosial yang mendalam bagi masyarakat manusia.
Revolusi "omics"
Omics neologisme berbahasa Inggris secara informal mengacu pada bidang studi di biologi yang diakhiri dengan -omics , seperti genomik, proteomik, atau metabolomik. Sufiks terkait -ome digunakan untuk menyebut objek studi pada bidang-bidang tersebut, seperti genom, proteome, atau metabolom masing-masing. Sufiks -ome yang digunakan dalam biologi molekuler mengacu pada totalitas dari beberapa jenis; Omics serupa telah merujuk secara umum pada studi kumpulan data biologis yang besar dan komprehensif. Sementara pertumbuhan penggunaan istilah telah menyebabkan beberapa ilmuwan (Jonathan Eisen, antara lain) mengklaim bahwa istilah tersebut telah terjual berlebihan, hal itu mencerminkan perubahan orientasi ke arah analisis kuantitatif dari bermacam-macam lengkap atau hampir lengkap dari semua konstituen dari sebuah sistem. Dalam studi simbiosis, misalnya, para peneliti yang dulunya hanya mempelajari produk gen tunggal sekarang dapat secara bersamaan membandingkan total komplemen dari beberapa jenis molekul biologis.
Analisis genom
Setelah organisme dipilih, proyek genom melibatkan tiga komponen: sekuensing DNA, perakitan sekuens itu untuk membuat representasi dari kromosom asli, dan anotasi serta analisis representasi itu.
Mengurutkan
Secara historis, pengurutan dilakukan di pusat pengurutan , fasilitas terpusat (mulai dari institusi independen besar seperti Joint Genome Institute yang mengurutkan puluhan terabase setahun, hingga fasilitas inti biologi molekuler lokal) yang berisi penelitian laboratorium dengan instrumentasi mahal dan dukungan teknis yang diperlukan. Namun, karena teknologi pengurutan terus meningkat, generasi baru pengurutan benchtop perputaran cepat yang efektif telah berada dalam jangkauan laboratorium akademik rata-rata. Secara keseluruhan, pendekatan pengurutan genom terbagi dalam dua kategori besar, pengurutan shotgun dan throughput tinggi (atau generasi berikutnya ).
Pengurutan senapan adalah metode pengurutan yang dirancang untuk analisis rangkaian DNA yang lebih panjang dari 1000 pasangan basa, hingga dan termasuk seluruh kromosom. Dinamai dengan analogi dengan pola penembakan semu acak yang berkembang pesat. Karena urutan elektroforesis gel hanya dapat digunakan untuk urutan yang cukup pendek (100 hingga 1000 pasangan basa), urutan DNA yang lebih panjang harus dipecah menjadi segmen kecil acak yang kemudian diurutkan untuk mendapatkan pembacaan . Beberapa pembacaan yang tumpang tindih untuk DNA target diperoleh dengan melakukan beberapa putaran fragmentasi dan sekuensing ini. Program komputer kemudian menggunakan ujung yang tumpang tindih dari bacaan yang berbeda untuk merangkainya menjadi urutan yang berkelanjutan. Urutan senapan adalah proses pengambilan sampel acak, memerlukan pengambilan sampel berlebih untuk memastikan nukleotida tertentu terwakili dalam urutan yang direkonstruksi; jumlah rata-rata pembacaan di mana genom diambil sampel berlebih disebut sebagai cakupan.
Untuk sebagian besar sejarahnya, teknologi yang mendasari pengurutan senapan adalah metode penghentian rantai klasik atau 'metode Sanger', yang didasarkan pada penggabungan selektif dari pemutusan rantai dideoxynucleotides oleh DNA polimerase selama replikasi DNA in vitro. Baru-baru ini, pengurutan senapan telah digantikan oleh metode pengurutan throughput tinggi, terutama untuk analisis genom otomatis berskala besar. Namun, metode Sanger tetap digunakan secara luas, terutama untuk proyek skala kecil dan untuk mendapatkan pembacaan sekuens DNA yang berdekatan (& gt; 500 nukleotida). Metode penghentian rantai memerlukan templat DNA untai tunggal, primer DNA, polimerase DNA, deoxynucleosidetriphosphates (dNTPs) normal, dan nukleotida termodifikasi (dideoxyNTPs) yang menghentikan pemanjangan untai DNA. Nukleotida pemutusan rantai ini kekurangan gugus 3'-OH yang diperlukan untuk pembentukan ikatan fosfodiester antara dua nukleotida, menyebabkan DNA polimerase menghentikan ekstensi DNA ketika ddNTP digabungkan. DdNTPs mungkin diberi label radioaktif atau fluoresen untuk deteksi dalam pengurut DNA. Biasanya, mesin ini dapat mengurutkan hingga 96 sampel DNA dalam satu batch (berjalan) hingga 48 kali sehari.
Permintaan yang tinggi untuk pengurutan berbiaya rendah telah mendorong pengembangan pengurutan throughput tinggi teknologi yang memparalelkan proses pengurutan, menghasilkan ribuan atau jutaan urutan sekaligus. Pengurutan throughput tinggi dimaksudkan untuk menurunkan biaya pengurutan DNA melebihi apa yang mungkin dilakukan dengan metode pewarna-terminator standar. Dalam pengurutan throughput yang sangat tinggi, sebanyak 500.000 operasi pengurutan demi sintesis dapat dijalankan secara paralel.
Metode pengurutan pewarna Illumina didasarkan pada terminator pewarna yang dapat dibalik dan dikembangkan pada tahun 1996 di Institut Penelitian Biomedis Jenewa, oleh Pascal Mayer dan Laurent Farinelli. Dalam metode ini, molekul DNA dan primer pertama kali dilampirkan pada slide dan diperkuat dengan polimerase sehingga koloni klonal lokal, yang pada awalnya disebut "koloni DNA", terbentuk. Untuk menentukan urutannya, empat jenis basis terminator yang dapat dibalik (basis-RT) ditambahkan dan nukleotida yang tidak tergabung dicuci. Tidak seperti pyrosequencing, rantai DNA diperpanjang satu nukleotida pada satu waktu dan akuisisi gambar dapat dilakukan pada saat yang tertunda, memungkinkan susunan koloni DNA yang sangat besar untuk ditangkap oleh gambar berurutan yang diambil dari satu kamera. Memisahkan reaksi enzimatis dan pengambilan gambar memungkinkan throughput yang optimal dan kapasitas pengurutan yang secara teoritis tidak terbatas; dengan konfigurasi optimal, hasil akhir instrumen hanya bergantung pada tingkat konversi A / D kamera. Kamera mengambil gambar dari nukleotida berlabel fluoresen, kemudian pewarna bersama dengan penghambat terminal 3 'secara kimiawi dikeluarkan dari DNA, memungkinkan siklus berikutnya.
Pendekatan alternatif, pengurutan semikonduktor ion, didasarkan pada kimia replikasi DNA standar. Teknologi ini mengukur pelepasan ion hidrogen setiap kali basa digabungkan. Sebuah microwell yang mengandung DNA cetakan dibanjiri dengan satu nukleotida, jika nukleotida tersebut melengkapi untai cetakan itu akan digabungkan dan sebuah ion hidrogen akan dilepaskan. Rilis ini memicu sensor ion ISFET. Jika homopolimer ada dalam urutan template, beberapa nukleotida akan digabungkan dalam satu siklus banjir, dan sinyal listrik yang terdeteksi akan lebih tinggi secara proporsional.
Majelis
Rakitan urutan mengacu pada penyelarasan dan menggabungkan fragmen dari urutan DNA yang lebih panjang untuk merekonstruksi urutan aslinya. Ini diperlukan karena teknologi sekuensing DNA saat ini tidak dapat membaca seluruh genom sebagai sekuens kontinu, melainkan membaca potongan kecil antara 20 dan 1000 basis, tergantung pada teknologi yang digunakan. Teknologi pengurutan generasi ketiga seperti PacBio atau Oxford Nanopore secara rutin menghasilkan pembacaan pengurutan & gt; 10 kb panjangnya; namun, mereka memiliki tingkat kesalahan yang tinggi sekitar 15 persen. Biasanya fragmen pendek, yang disebut bacaan, hasil dari DNA genomik pengurutan senapan, atau transkrip gen (EST).
Rakitan secara luas dapat dikategorikan menjadi dua pendekatan: perakitan de novo , untuk genom yang tidak mirip dengan urutan mana pun di masa lalu, dan perakitan komparatif, yang menggunakan urutan yang ada dari organisme terkait erat sebagai referensi selama perakitan. Sehubungan dengan perakitan komparatif, perakitan de novo sulit secara komputasi (NP-hard), sehingga kurang disukai untuk teknologi NGS baca singkat. Dalam paradigma perakitan de novo ada dua strategi utama untuk perakitan, strategi jalur Eulerian, dan strategi konsensus-tata letak-tumpang tindih (OLC). Strategi OLC pada akhirnya mencoba membuat jalur Hamiltonian melalui grafik tumpang tindih yang merupakan masalah NP-hard. Strategi jalur Eulerian secara komputasi lebih mudah diatur karena mereka mencoba menemukan jalur Euler melalui grafik deBruijn.
Genom yang sudah selesai didefinisikan sebagai memiliki satu urutan yang berdekatan tanpa ambiguitas yang mewakili setiap replikon.
Anotasi
Rakitan urutan DNA saja memiliki nilai kecil tanpa analisis tambahan. Anotasi genom adalah proses melampirkan informasi biologis ke urutan, dan terdiri dari tiga langkah utama:
- mengidentifikasi bagian genom yang tidak mengkode protein
- mengidentifikasi elemen pada genom, proses yang disebut prediksi gen, dan
- melampirkan informasi biologis ke elemen-elemen ini.
Alat anotasi otomatis mencoba melakukan langkah-langkah berikut in silico , sebagai lawan dari anotasi manual (alias kurasi) yang melibatkan keahlian manusia dan verifikasi eksperimental potensial. Idealnya, pendekatan ini hidup berdampingan dan saling melengkapi dalam saluran anotasi yang sama (juga lihat di bawah).
Secara tradisional, tingkat dasar anotasi menggunakan BLAST untuk menemukan kesamaan, dan kemudian membuat anotasi genom berdasarkan homolog . Baru-baru ini, informasi tambahan ditambahkan ke platform anotasi. Informasi tambahan memungkinkan anotator manual untuk mendekonvolusi perbedaan antara gen yang diberi anotasi yang sama. Beberapa database menggunakan informasi konteks genom, skor kemiripan, data eksperimen, dan integrasi sumber daya lain untuk memberikan penjelasan genom melalui pendekatan Subsistem mereka. Basis data lain (misalnya Ensembl) mengandalkan sumber data yang dikurasi serta berbagai alat perangkat lunak dalam saluran anotasi genom otomatis mereka. Anotasi struktural terdiri dari identifikasi elemen genom, terutama ORF dan lokalisasinya, atau struktur gen. Anotasi fungsional terdiri dari melampirkan informasi biologis ke elemen genomik.
Mengurutkan pipeline dan database
Perlunya reproduktifitas dan pengelolaan yang efisien dari sejumlah besar data yang terkait dengan proyek genom berarti pipeline komputasi memiliki aplikasi penting dalam genomik.
Area penelitian
Genomik fungsional
Genomik fungsional adalah bidang biologi molekuler yang mencoba memanfaatkan begitu banyak data yang dihasilkan oleh proyek genom (seperti proyek pengurutan genom) untuk mendeskripsikan fungsi dan interaksi gen (dan protein). Genomik fungsional berfokus pada aspek dinamis seperti transkripsi gen, terjemahan, dan interaksi protein-protein, yang bertentangan dengan aspek statis informasi genom seperti urutan atau struktur DNA. Genomik fungsional mencoba menjawab pertanyaan tentang fungsi DNA pada tingkat gen, transkrip RNA, dan produk protein. Karakteristik utama studi genomik fungsional adalah pendekatan seluruh genom mereka terhadap pertanyaan-pertanyaan ini, umumnya melibatkan metode throughput tinggi daripada pendekatan “gen-demi-gen” yang lebih tradisional.
Cabang utama genomik adalah masih peduli dengan sekuensing genom berbagai organisme, tetapi pengetahuan tentang genom lengkap telah menciptakan kemungkinan untuk bidang genomik fungsional, terutama berkaitan dengan pola ekspresi gen selama berbagai kondisi. Alat yang paling penting di sini adalah mikroarray dan bioinformatika.
Genomik struktural
Genomik struktural berupaya mendeskripsikan struktur 3 dimensi dari setiap protein yang dikodekan oleh genom tertentu. Pendekatan berbasis genom ini memungkinkan metode penentuan struktur dengan hasil yang tinggi melalui kombinasi pendekatan eksperimental dan pemodelan. Perbedaan utama antara genomik struktural dan prediksi struktural tradisional adalah bahwa genomik struktural mencoba menentukan struktur setiap protein yang dikodekan oleh genom, daripada berfokus pada satu protein tertentu. Dengan tersedianya sekuens genom lengkap, prediksi struktur dapat dilakukan lebih cepat melalui kombinasi pendekatan eksperimental dan pemodelan, terutama karena ketersediaan sejumlah besar sekuens genom dan struktur protein terpecahkan sebelumnya memungkinkan para ilmuwan untuk memodelkan struktur protein pada struktur yang dipecahkan sebelumnya. homolog. Genomik struktural melibatkan pengambilan sejumlah besar pendekatan untuk penentuan struktur, termasuk metode eksperimental menggunakan urutan genomik atau pendekatan berbasis pemodelan berdasarkan urutan atau homologi struktural ke protein dengan struktur yang diketahui atau berdasarkan prinsip kimia dan fisika untuk protein tanpa homologi untuk struktur apa pun yang diketahui. Berbeda dengan biologi struktural tradisional, penentuan struktur protein melalui upaya genomik struktural sering kali (tetapi tidak selalu) datang sebelum apa pun yang diketahui tentang fungsi protein. Hal ini menimbulkan tantangan baru dalam bioinformatika struktural, yaitu menentukan fungsi protein dari struktur 3D-nya.
Epigenomik
Epigenomik adalah studi tentang rangkaian lengkap modifikasi epigenetik pada materi genetik sebuah sel. , dikenal sebagai epigenom. Modifikasi epigenetik adalah modifikasi reversibel pada DNA sel atau histon yang mempengaruhi ekspresi gen tanpa mengubah urutan DNA (Russell 2010 p. 475). Dua dari modifikasi epigenetik yang paling khas adalah metilasi DNA dan modifikasi histon. Modifikasi epigenetik memainkan peran penting dalam ekspresi dan regulasi gen, dan terlibat dalam berbagai proses seluler seperti diferensiasi / perkembangan dan tumorigenesis. Studi tentang epigenetik di tingkat global baru-baru ini dimungkinkan melalui adaptasi uji throughput tinggi genomik.
Metagenomik
Metagenomik adalah studi tentang metagenom , materi genetik yang diperoleh langsung dari sampel lingkungan. Bidang luas juga dapat disebut sebagai genomik lingkungan, ekogenomik atau genomik komunitas. Sementara mikrobiologi tradisional dan sekuensing genom mikroba bergantung pada kultur klonal yang dibudidayakan, sekuensing gen lingkungan awal kloning gen spesifik (seringkali gen 16S rRNA) untuk menghasilkan profil keragaman dalam sampel alami. Pekerjaan tersebut mengungkapkan bahwa sebagian besar keanekaragaman hayati mikroba telah hilang dengan metode berbasis budidaya. Studi terbaru menggunakan sekuensing Sanger "senapan" atau pyrosequencing paralel besar-besaran untuk mendapatkan sampel yang sebagian besar tidak bias dari semua gen dari semua anggota komunitas sampel. Karena kemampuannya untuk mengungkap keragaman kehidupan mikroskopis yang sebelumnya tersembunyi, metagenomik menawarkan lensa canggih untuk melihat dunia mikroba yang berpotensi merevolusi pemahaman tentang seluruh dunia kehidupan.
Sistem model
Bakteriofag telah memainkan dan terus memainkan peran kunci dalam genetika bakteri dan biologi molekuler. Secara historis, mereka digunakan untuk mendefinisikan struktur gen dan regulasi gen. Juga genom pertama yang diurutkan adalah bakteriofag. Namun, penelitian bakteriofag tidak memimpin revolusi genomik, yang jelas-jelas didominasi oleh genomik bakteri. Baru belakangan ini studi tentang genom bakteriofag menjadi menonjol, sehingga memungkinkan para peneliti untuk memahami mekanisme yang mendasari evolusi fag. Urutan genom bakteriofag dapat diperoleh melalui sekuensing langsung dari bakteriofag yang diisolasi, tetapi juga dapat diturunkan sebagai bagian dari genom mikroba. Analisis genom bakteri telah menunjukkan bahwa sejumlah besar DNA mikroba terdiri dari urutan profag dan elemen mirip profag. Penambangan basis data terperinci dari sekuens ini menawarkan wawasan tentang peran profag dalam membentuk genom bakteri: Secara keseluruhan, metode ini memverifikasi banyak kelompok bakteriofag yang dikenal, menjadikannya alat yang berguna untuk memprediksi hubungan profag dari genom bakteri.
Saat ini ada 24 cyanobacteria yang total urutan genomnya tersedia. 15 dari cyanobacteria ini berasal dari lingkungan laut. Ini adalah enam strain Prochlorococcus , tujuh strain Synechococcus laut, Trichodesmium erythraeum IMS101 dan Crocosphaera watsonii WH8501. Beberapa penelitian telah menunjukkan bagaimana urutan ini dapat digunakan dengan sangat berhasil untuk menyimpulkan karakteristik ekologi dan fisiologis penting dari cyanobacteria laut. Namun, masih banyak lagi proyek genom yang sedang berlangsung, di antaranya terdapat isolasi Prochlorococcus dan laut Synechococcus , Acaryochloris dan Prochloron. , cyanobacteria berfilamen pengikat N2 Nodularia spumigena , Lyngbya aestuarii dan Lyngbya majuscula , serta bakteriofag yang menginfeksi cyanobaceria laut. Dengan demikian, informasi genom yang terus berkembang juga dapat dimanfaatkan dengan cara yang lebih umum untuk mengatasi masalah global dengan menerapkan pendekatan komparatif. Beberapa contoh kemajuan baru dan menarik dalam bidang ini adalah identifikasi gen untuk RNA pengatur, wawasan tentang asal mula evolusi fotosintesis, atau perkiraan kontribusi transfer gen horizontal ke genom yang telah dianalisis.
Aplikasi genomik
Genomik telah menyediakan aplikasi di banyak bidang, termasuk kedokteran, bioteknologi, antropologi, dan ilmu sosial lainnya.
Pengobatan genomik
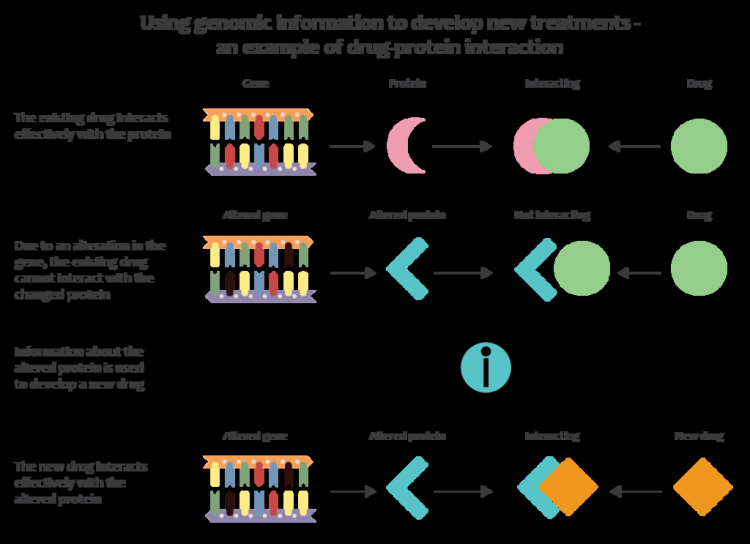
Genomik generasi selanjutnya Teknologi memungkinkan dokter dan peneliti biomedis untuk secara drastis meningkatkan jumlah data genom yang dikumpulkan pada populasi penelitian yang besar. Ketika dikombinasikan dengan pendekatan informatika baru yang mengintegrasikan banyak jenis data dengan data genomik dalam penelitian penyakit, hal ini memungkinkan peneliti untuk lebih memahami basis genetik dari respon obat dan penyakit. Upaya awal untuk menerapkan genom pada pengobatan termasuk yang dilakukan oleh tim Stanford yang dipimpin oleh Euan Ashley yang mengembangkan alat pertama untuk interpretasi medis dari genom manusia. Misalnya, program penelitian Kita Semua bertujuan untuk mengumpulkan data urutan genom dari 1 juta peserta untuk menjadi komponen penting dari platform penelitian kedokteran presisi.
Biologi sintetik dan bioteknologi
Pertumbuhan pengetahuan genom telah memungkinkan penerapan biologi sintetik yang semakin canggih. Pada tahun 2010, para peneliti di J. Craig Venter Institute mengumumkan pembuatan spesies bakteri yang sebagian sintetik, laboratorium Mycoplasma , yang diturunkan dari genom Mycoplasma genitalium .
Genomik konservasi
Konservasionis dapat menggunakan informasi yang dikumpulkan oleh pengurutan genom untuk mengevaluasi dengan lebih baik faktor genetik kunci konservasi spesies, seperti keragaman genetik suatu populasi atau apakah seseorang heterozigot untuk resesif kelainan genetik yang diturunkan. Dengan menggunakan data genom untuk mengevaluasi efek proses evolusi dan untuk mendeteksi pola dalam variasi di seluruh populasi tertentu, ahli konservasi dapat merumuskan rencana untuk membantu spesies tertentu tanpa banyak variabel yang tidak diketahui seperti yang tidak ditangani oleh pendekatan genetik standar.
Gugi Health: Improve your health, one day at a time!

Genetika Mungkin Tidak Banyak Berhubungan Dengan Harapan Hidup Anda — Tapi Ini Ada
Berapa lama kita hidup tampaknya tidak terlalu berkaitan dengan gen kita …

George W. Citroner
George W. Citroner meliput berita terbaru di bidang kedokteran dan kesehatan. …
